An Invert Relationship between the Expression of MnSOD and p53

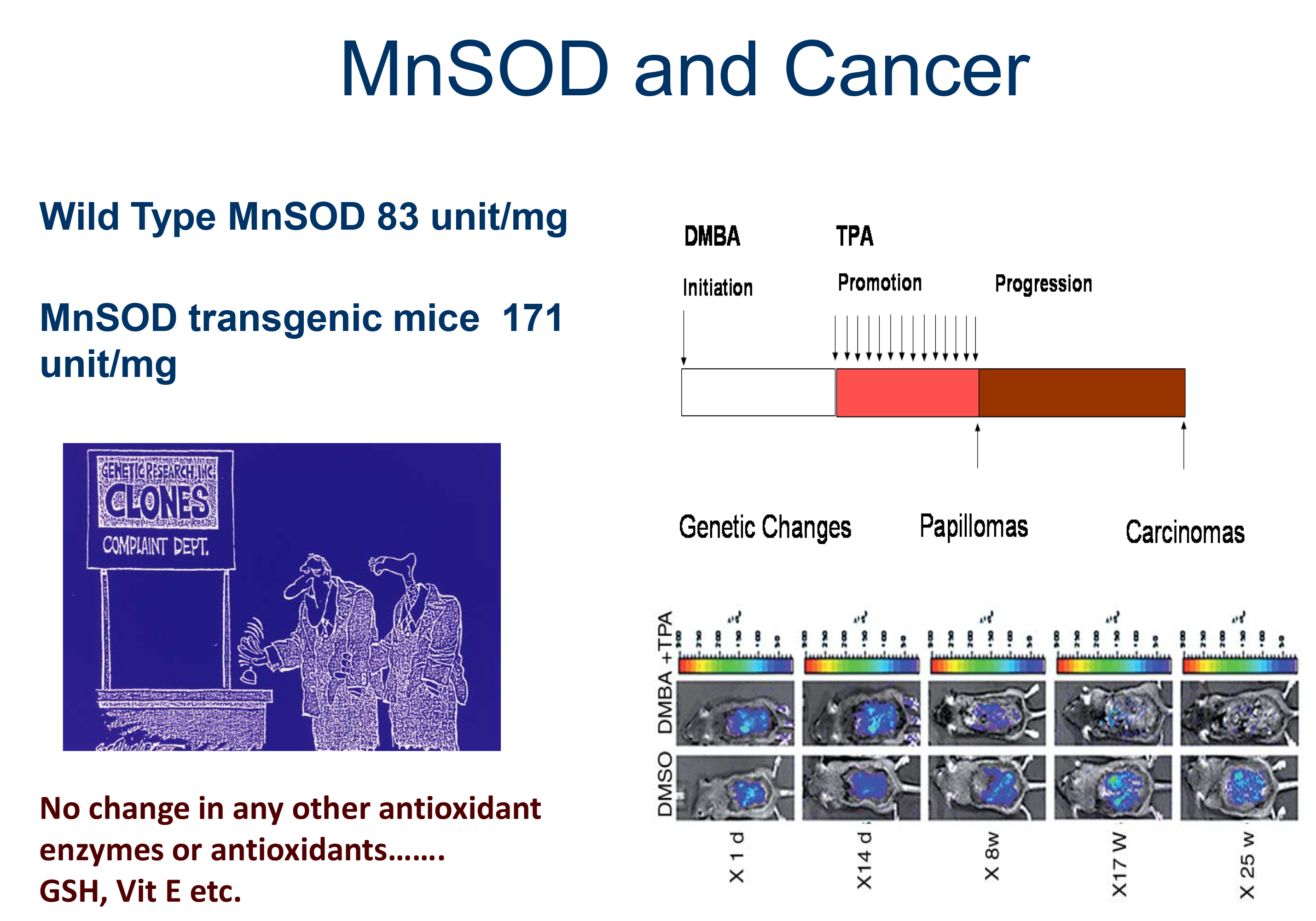
MnSOD and Cancer
Researching the mitochondrial antioxidant defense system
Find Out MoreOur overreaching goal is to develop an approach to combination therapy that will increase the potential for cancer patients to survive and to improve their quality of life. Our emphasis is on the development of a dual-purpose drug that will: 1) improve the efficacy of existing cancer therapeutic protocols and 2) reduce the toxic side effects to normal tissues caused by existing therapeutic strategies. We expect that dual-purpose therapeutics will be the successful cancer treatment of the future.
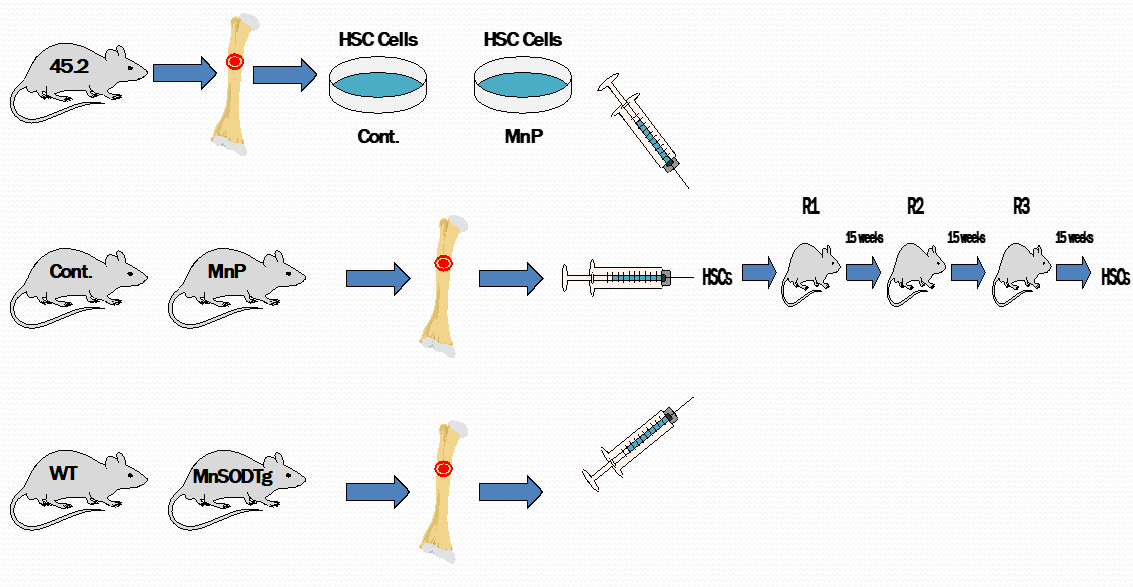
The primary research focus of this laboratory is in the area of the mitochondrial antioxidant defense system. We cloned the human gene for the primary superoxide removal enzyme in mitochondria, manganese superoxide dismutase (MnSOD), and the initial study has been expanded into several separate but related projects. These projects involve evaluating genetic abnormalities of antioxidant enzymes, the mechanisms regulating gene expression, and the impact these alterations have on the ability of humans to cope with oxidative stress. We made the seminal observation that expression of MnSOD in mitochondria suppresses neoplastic transformation and the death of cancer cells, leading to a reduction in their tumorigenicity and metastatic capability. We then turned our attention to addressing the unresolved issue of whether and when MnSOD expression is altered in cancer. To directly address this important question, we generated transgenic mice expressing a luciferase reporter gene under the control of human MnSOD promoter/enhancer elements to investigate the changes of MnSOD transcription throughout the development of cancer. The results demonstrate that MnSOD expression is suppressed at a very early stage, but subsequently increases at late stages of carcinogenesis, and also identify MnSOD as a p53-regulated gene whose expression switches between early and advanced stages of cancer. We further demonstrated that MnSOD serves as a mitochondrial fidelity protein that protects the mitochondrial genome against oxidative stress-induced inactivation. These critical findings have led us to our current concentration, testing the hypothesis that defective MnSOD activity signals an adaptive response, leading to activation of a metabolic switch that initiates the Warburg effect, which is an important metabolic change that confers many growth and survival advantages to cancer cells.
Generation of reactive oxygen species (ROS) is a major mechanism responsible for the therapeutic effect of ionizing radiation and many chemotherapeutic drugs. Under physiological conditions, mitochondria produce the most ROS in the cell, and the critical primary antioxidant enzyme protecting mitochondria is manganese superoxide dismutase (MnSOD). A shift in cell redox status toward an oxidizing condition activates mitochondrial retrograde signaling leading to activation of adaptive response or cell death pathways. We have identified the 4HNE adduction of apoptosis inducing factor associated mitochondrion protein 2 (AIFm2), a p53 target gene, as a mediator of mitochondrial retrograde signaling under pathophysiological conditions. The native AIFm2 localizes to mitochondria whereas HNE-adducted AIFm2 localizes to nucleus, suggesting that HNE causes the translocation of AIFm2. HNE selectively modifies His174 and Cys187 of the AIFm2 protein. The HNE-adducted AIFm2 loses its enzymatic activity function but exhibits strong DNA binding affinity. These findings provide evidence linking HNE adduction of a mitochondrial protein with protein trafficking and reveal a novel function of oxidatively modified proteins in retrograde signaling.

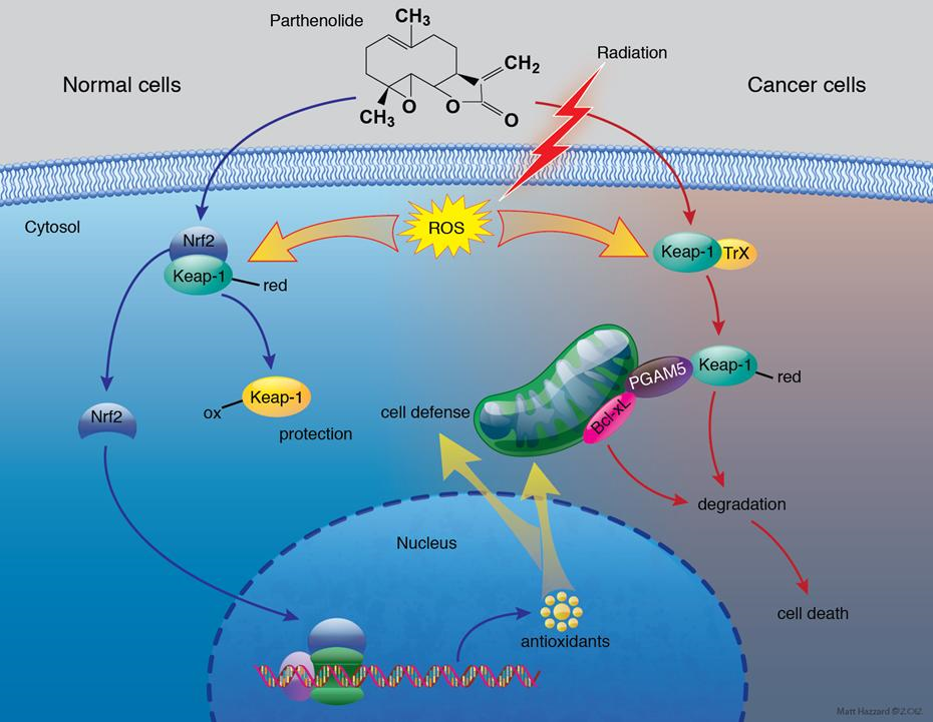
Generation of ROS is a major mechanism responsible for the therapeutic effect of ionizing radiation and nearly 50% of chemotherapeutic drugs. Currently, these therapeutic strategies are being used to kill cancer cells without the benefit of a rational design that exploits the intrinsic differences in the cellular redox status of normal cells and cancer cells. Cancer cells are usually under higher oxidative stress than normal cells and it is known that an additional increase in prooxidant level can trigger cell death. Thus, therapeutic approaches that use redox active antioxidants that push tumor cells into oxidative stress overload but stimulate adaptive responses in normal cells can be developed to selectively enhance the efficient killing of cancer cells by radiation/chemotherapy. We are developing a novel dual-purpose drug approach that would not only improve the efficacy of cancer therapy but could also improve quality of life for cancer survivors by protecting normal tissue from ROS-generating therapeutics.

Serious long-term therapy-induced impairment of cancer survivors is currently an understudied facet of cancer treatment, but one that is emerging as a critical need given clinical successes with certain cancers and treatment modalities. Our current research direction, based on in-depth understandings resulting from our decades-long focus, is into antioxidant defense mechanisms that have an intriguing potential to address the challenge of serious post-therapy impairment. Our team of investigators is engaged in a series of studies that move between bench and bedside to investigate a model that proposes that, in addition to the direct ROS-mediated mitochondrial injury in cardiac tissue, there is an indirect mechanism for drug-induced injury common to both cardiac and CNS tissues that results from an immune-mediated chain of events triggered by drug-induced oxidative modification of proteins. This team project is designed to identify practice- changing therapy through an integrated program of carefully coordinated mechanistic, intervention, and translational studies with the potential to limit currently devastating tissue toxicities that compromise critical physiological functions. Such advances may significantly improve the lives of cancer survivors.
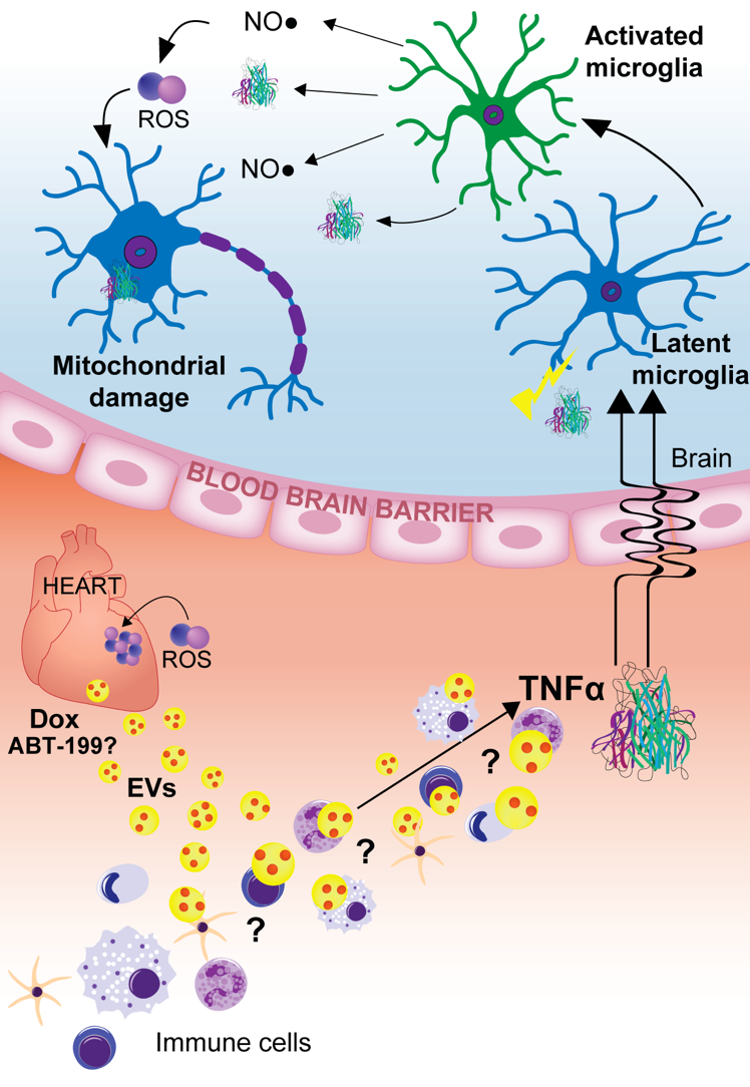
Yarana, C.;Carroll, D.;Chen, J.;Chaiswing, L.;Zhao, Y.;Noel, T.;Alstott, M.;Bae, Y.;Dressler, E.V.;Moscow, J.A.;Butterfield, D.A.;Zhu, H.;St Clair, D.K. "Extracellular vesicles released by cardiomyocytes in a doxorubicin-induced cardiac injury mouse model contain protein biomarkers of early cardiac injury." Clinical cancer research : an official journal of the American Association for Cancer Research (2017): [PubMed Link] | [ Full text ]
Wei, X.;Xu, Y.;Xu, F.F.;Chaiswing, L.;Schnell, D.;Noel, T.;Wang, C.;Chen, J.;St Clair, D.K.;St Clair, W.H. "RelB Expression Determines the Differential Effects of Ascorbic Acid in Normal and Cancer Cells." Cancer research 77, 6 (2017): 1345-1356. [PubMed Link] | [ Full text ]
Zhao, Y.;Miriyala, S.;Miao, L.;Mitov, M.;Schnell, D.;Dhar, S.K.;Cai, J.;Klein, J.B.;Sultana, R.;Butterfield, D.A.;Vore, M.;Batinic-Haberle, I.;Bondada, S.;St Clair, D.K. "Redox proteomic identification of HNE-bound mitochondrial proteins in cardiac tissues reveals a systemic effect on energy metabolism after doxorubicin treatment." Free radical biology & medicine 72, (2014): 55-65. [PubMed Link] | [ Full text ]
Sun, Y.;St Clair, D.K.;Xu, Y.;Crooks, P.A.;St Clair, W.H. "A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells." Cancer research 70, 7 (2010): 2880-90. [PubMed Link] | [ Full text ]
Xu, Y.;Josson, S.;Fang, F.;Oberley, T.D.;St Clair, D.K.;Wan, X.S.;Sun, Y.;Bakthavatchalu, V.;Muthuswamy, A.;St Clair, W.H. "RelB enhances prostate cancer growth: implications for the role of the nuclear factor-kappaB alternative pathway in tumorigenicity." Cancer research 69, 8 (2009): 3267-71. [PubMed Link] | [ Full text ]
Lien, Y.C.;Noel, T.;Liu, H.;Stromberg, A.J.;Chen, K.C.;St Clair, D.K. "Phospholipase C-delta1 is a critical target for tumor necrosis factor receptor-mediated protection against adriamycin-induced cardiac injury." Cancer research 66, 8 (2006): 4329-38. [PubMed Link] | [ Full text ]
Yen, H.C.;Oberley, T.D.;Vichitbandha, S.;Ho, Y.S.;St Clair, D.K. "The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice." The Journal of clinical investigation 98, 5 (1996): 1253-60. [PubMed Link] | [ Full text ]
Urano, M.;Kuroda, M.;Reynolds, R.;Oberley, T.D.;St Clair, D.K. "Expression of manganese superoxide dismutase reduces tumor control radiation dose: gene-radiotherapy." Cancer research 55, 12 (1995): 2490-3. [PubMed Link] | [ Full text ]
St Clair, D.K.;Wan, X.S.;Oberley, T.D.;Muse, K.E.;St Clair, W.H. "Suppression of radiation-induced neoplastic transformation by overexpression of mitochondrial superoxide dismutase." Molecular carcinogenesis 6, 4 (1992): 238-42. [PubMed Link] | [ Full text ]
Toxicology and Cancer Biology
University of Kentucky
454 Bosomworth HSRB
1095 VA Drive
Lexington, KY 40536